· Samantha J. Klasfeld, Ph.D. · Tutorial ·5 min read · Original Source
Biobank Intro Series: Hardware Settings
Hardware setup lessons for UK Biobank Research Analysis Platform and All of Us Workbench
In 2026 the most expensive resource isn’t compute time or storage. It’s your time rerunning failed analyses. After months of working across the UK Biobank RAP and All of Us Researcher Workbench, I’ve collected some hard-won lessons about resource management.
This post is more technical. Consider yourself warned.
Tip #1: Each Platform Has Its Own Command-Line Interface (CLI)
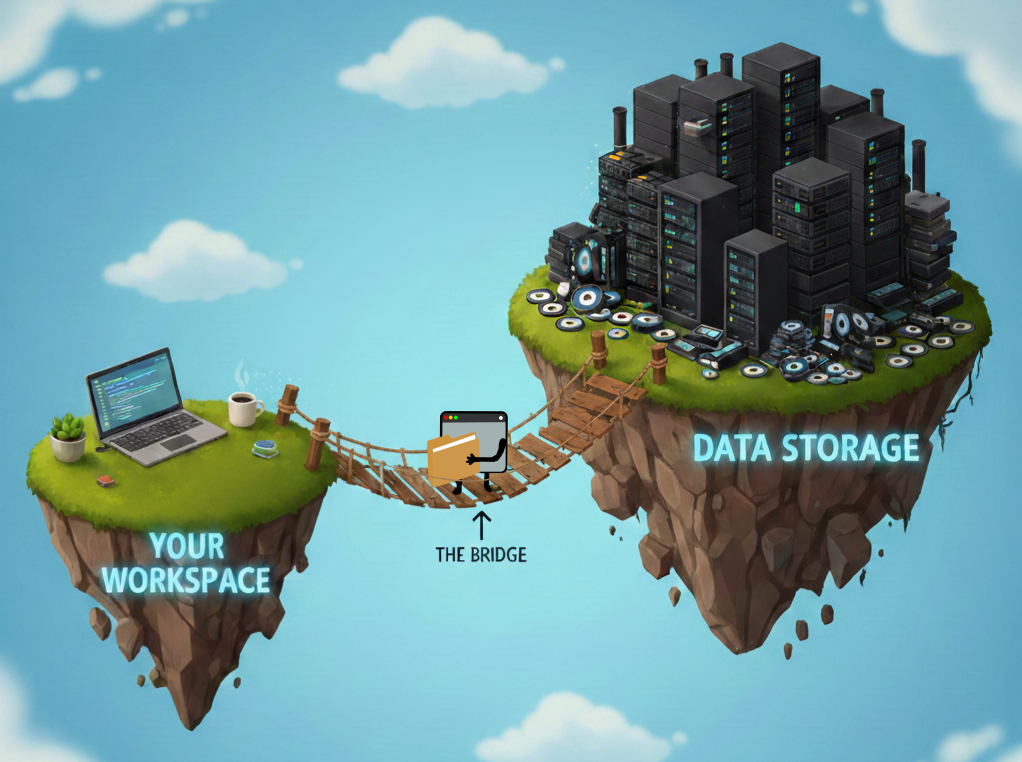
Think of your workspace and the platform’s data storage as two separate floating islands. Your code lives on one island. The massive biobank files live on the other. The CLI is the bridge between them, and each platform has its own.
UK Biobank RAP: The dx toolkit
The dx CLI is your friend for navigating the RAP filesystem:
# List files in a directory within the platform's data storage
dx ls
# Stream file contents (don't download!) from data storage
dx cat file-xxxx | bcftools view
# Upload local files from your workspace to data storage
dx upload local_file.txt --path /file/path/in/workspace/
# Download files (only if absolutely necessary) to your workspace
dx download file-xxxx --output local_file.txtAll of Us: gsutil for Google Cloud Storage
All of Us data lives in Google Cloud Storage (GCS) buckets and uses gsutil to identify, stream, and move data between your workspace and these buckets.
# List files in the controlled data-repository (CDR) bucket
gsutil ls gs://fc-aou-datasets-controlled/
# Find VCF files in the CDR bucket
gsutil ls gs://fc-aou-datasets-controlled/v7/wgs/short_read/snpindel/
# Stream directly (the right way)
gsutil cat gs://path/to/file.vcf.gz | bcftools viewImportant: Add a -u flag to gsutil commands to attribute the operation to your project for proper billing and access control:
gsutil -u $GOOGLE_PROJECT [command]Use the environment variables $GOOGLE_PROJECT and $WORKSPACE_BUCKET to avoid hardcoding paths:
# Upload local file from your workspace to your storage bucket
gsutil -u $GOOGLE_PROJECT cp local_file.txt $WORKSPACE_BUCKET/
# Download files (only if absolutely necessary) to your workspace
gsutil -u $GOOGLE_PROJECT cp gs://path/to/file.txt local_file.txtTip #2: Don’t Bring the Cloud Home With You
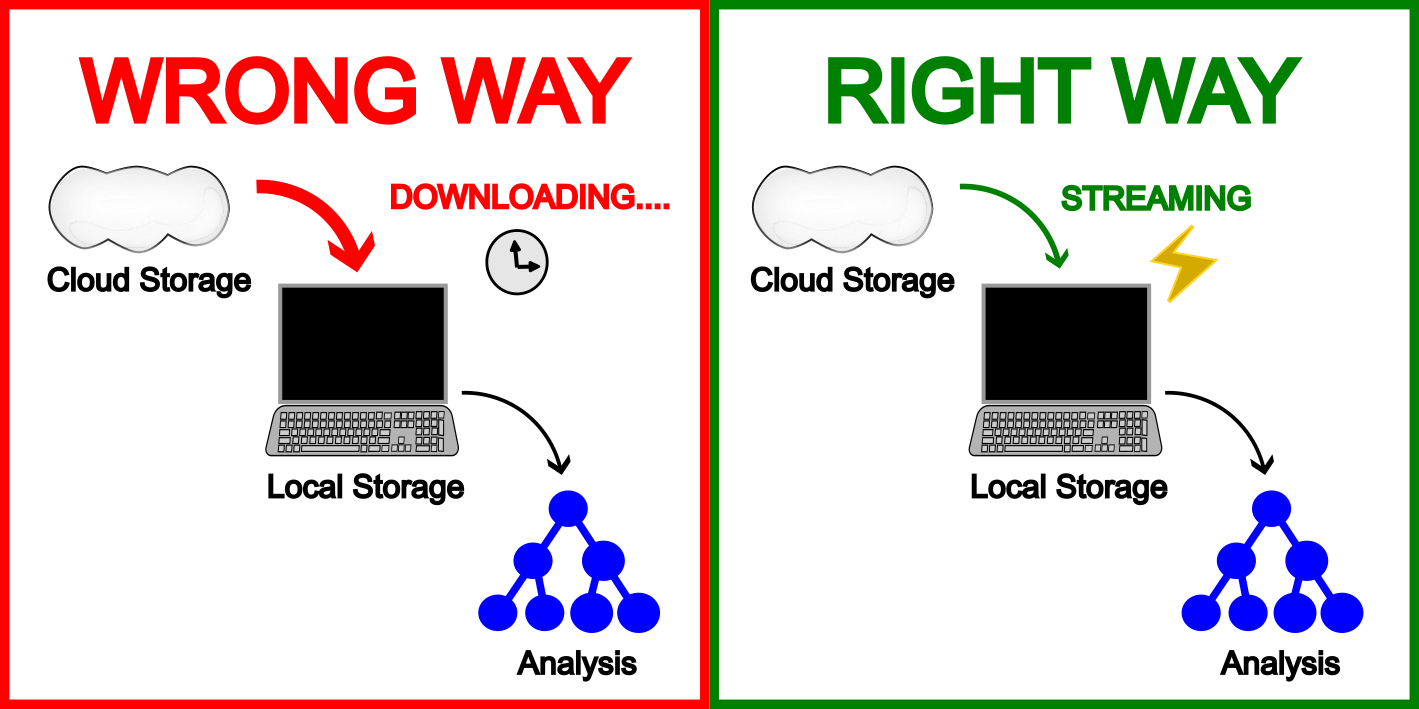
Now that you know how to upload and download files, I must restate that you should not use those download commands on biobank data files. Yes, dx download and gsutil cp exist, but the data is already where it needs to be. Your job is to meet it there, not drag it to you.
Don’t do this:
# UK Biobank: Copying a 500GB VCF locally
dx download file-xxxx
# All of Us: Same mistake, different platform
gsutil cp gs://path/to/huge.vcf.gz .Do this instead:
# UK Biobank: Stream with dx
dx cat file-xxxx | bcftools view | your_analysis
# All of Us: Stream with gsutil
gsutil cat gs://path/to/file.vcf.gz | bcftools view | your_analysisThe data is already where it needs to be. It sits in the cloud, on fast storage, ready to be streamed. Copying wastes time, burns through storage quotas, and risks running out of disk space mid-analysis. Both platforms are designed for streaming access. Use it.
Tip #3: Know Your Tools and Your Files
Hail is prominently featured in All of Us documentation, which makes it tempting to reach for first. Resist that instinct and match your tool to your actual problem, not the first one you find or the most impressive-sounding one.
Why avoid Hail when possible:
- Requires expensive Spark clusters
- Memory-intensive operations that crash your instance
- Adds complexity when simpler tools work fine
If you can use standard tools (pandas, bcftools, plink), do that instead. Save Hail for genuinely distributed computing tasks.
Regardless of which tool you choose, make sure your files are indexed (.tbi, .csi) before querying them. Without an index, tools like bcftools have to read the entire file from start to finish — region queries are no faster than loading everything.
Tip #4: Don’t Let Long Jobs Catch You Off Guard
Picture this: You start a 2-hour variant annotation job, grab lunch, and return to… nothing. Just a terminated instance.
Three habits will save you from rerunning everything from scratch.
First, check your idle timeout limit before running any long job. By default, All of Us shuts down after 15 minutes of inactivity.
- All of Us: Go to workspace settings → increase idle timeout to 8 hours (or your preferred duration)
- UK Biobank RAP: Check instance auto-pause settings
Note: nohup or screen/tmux can keep jobs running but won’t survive an instance shutdown so adjusting your timeout is still necessary.
Second, filter as early in your pipeline as possible. The less data you’re carrying through each step, the faster and cheaper each step is.
Third, checkpoint intermediate results. Save outputs at meaningful stages so a crash at step 5 doesn’t send you back to step 1.
None of these take more than a minute to set up. The rerun will.
The Bottom Line
When things crash:
- You ran out of memory
- Filter earlier in your pipeline
- Checkpoint intermediate results
When things are slow:
- Check if files are indexed (.tbi, .csi)
- Use region queries instead of full chromosomes
- Stream instead of copying
Set yourself up for success: learn your CLIs, stream your data, pick the right tool for the job, and make sure your long jobs have safety nets.