· Samantha J. Klasfeld, Ph.D. · Tutorial ·7 min read · Original Source
Biobank Intro Series: UK Biobank Genetic Data
Traversing genetic data on the UK Biobank RAP (Research Analysis Platform) environment
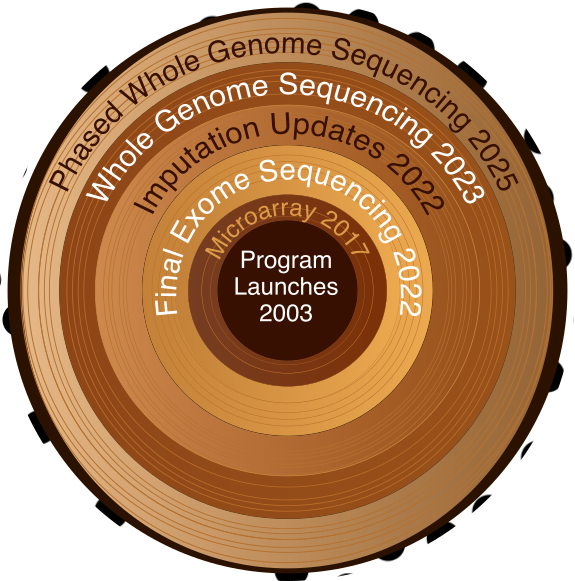
UK Biobank's genetic data has grown over time, from microarray genotyping to whole genome sequencing.
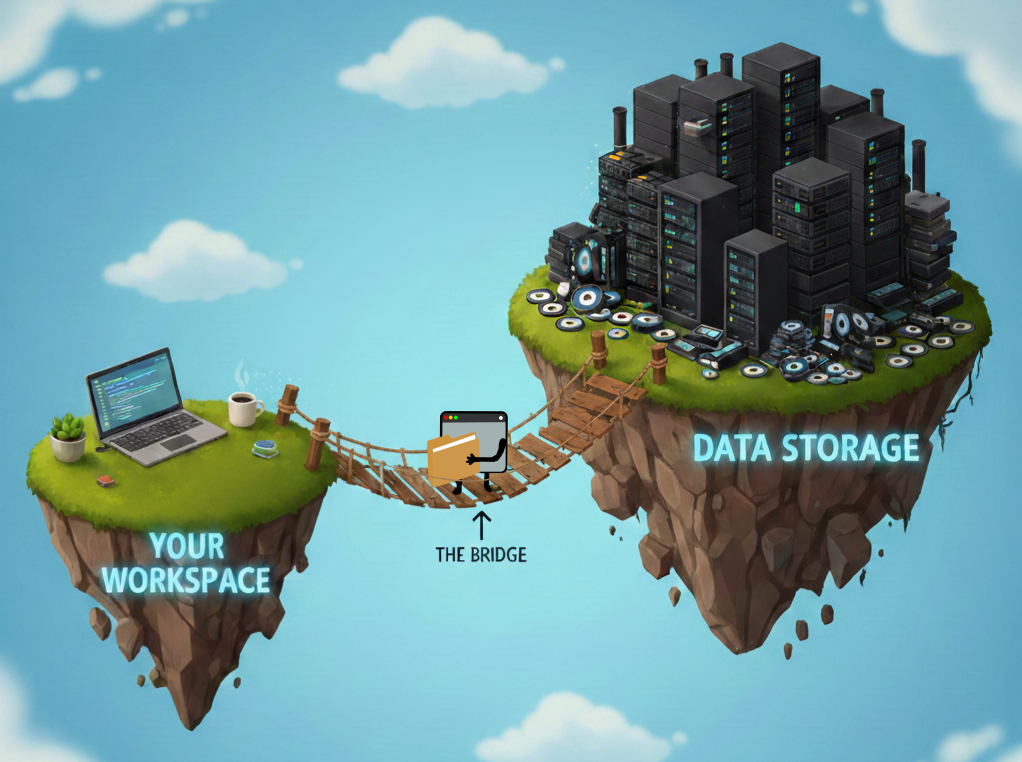
Genetic data. We made it. Coming from my Ph.D. in plant epigenetics, I was used to raw sequencing data from one replicate at a time. In contrast, the UK Biobank (UKB) has around 500,000 participants. The enormity of it can become an unbearable dead weight if you let it. The fix is straightforward: fit the data to your analysis, not the other way around. In my work, I’ve focused on a single region or a handful of variants at a time. That means streaming raw files from the data repository and filtering on the fly, rather than pulling them down in full with DNAnexus (see my previous blog on UKB hardware). The first step is knowing what genetic data UKB actually has, where it lives, and how to filter it before it becomes your problem.
The Showcase Knows About Genetics Too
In my previous post about the UKB Showcase, I focused on observational fields, but the Showcase also catalogs genetic data resources, including:
- Microarray genotyping data — directly measured variants at selected positions across the genome
- Imputed genotypes — variants inferred by combining array data with full-genome reference panels
- Exome sequencing data — sequenced protein-coding regions (~2% of the genome)
- Whole genome sequencing — full genome coverage
- Phased haplotypes — estimates of which alleles sit on the same chromosome (i.e., are inherited together)
Depending on the research question, you may need multiple data types. For example, phased haplotype data provides haplotype structure but excludes rare variants, so you might pair it with whole genome sequencing calls to get both. These data types were generated using different tools and released at different times, and I find it helpful to know that context to not only pick the right data, but also to understand the publications that used UKB genetic data before me. UKB provides a comprehensive timeline of their data releases, but coming from a sequencing background, the terminology didn’t sit well with me. For example, “genotyping data” actually means microarray data — not sequencing. Here are the current releases you’ll most likely use:
| Year | Data Type | Field ID | Participants | Notes |
|---|---|---|---|---|
| 2017 | Microarray + imputation | 22418 | 488,377 | ~800K measured; ~96M imputed 1 |
| 2022 | Exome sequencing (final) | 23141 | ~470,000 | |
| 2022 | Imputation (new panels) | 21007 | 488,377 | GEL and TOPMed; now in GRCh38 |
| 2023 | Whole genome sequencing | 24311 | ~500,000 | ML corrections; DRAGEN; unphased |
| 2025 | WGS (updated) | 30108 | ~500,000 | phased VCFs |
And here are the earlier releases you may encounter in older publications:
| Year | Data Type | Participants | Notes |
|---|---|---|---|
| 2019 | Exome sequencing | 50,000 | Alignment errors identified 2 |
| 2020 | Exome sequencing (reprocessed) | 200,000 | New OQFE pipeline 3 |
| 2021 | Exome sequencing | ~454,000 | 4 |
| 2021 | Whole genome sequencing | 200,000 | 5 |
Where Are the Files? (The Practical Part)
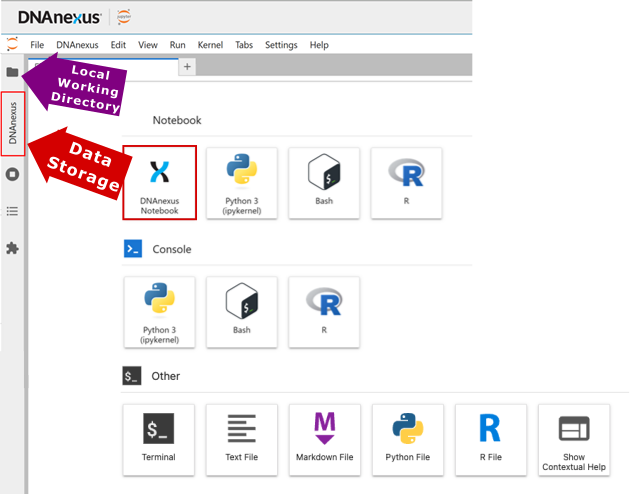
If you need a refresher on the two storage spaces, see the hardware post. The genetic data lives in data storage under a directory called “Bulk”. Filenames contain field ID numbers, so you can cross-reference them with the UK Biobank Showcase. For WGS data, field ID 24311 corresponds to the ML-corrected DRAGEN release. As of the latest release, this is often the one you want.
Start broad to learn the file structure, then narrow down. Running
dx find data --name "ukb24311*.vcf.gz" --folder /Bulk | headreveals that files follow the pattern:
/Bulk/DRAGEN WGS/ML-corrected DRAGEN population level WGS variants, pVCF format [500k release]/chr{CHROMOSOME}/ukb24311_c{CHROMOSOME}_b{BATCH}_v1.vcf.gz
Yes, the path has spaces. Always quote it. Once you know that, you can narrow the search to a specific chromosome. Even then, WGS data is split across many batch files, so | head is still warranted:
dx find data --name "ukb24311_c11_*.vcf.gz" --folder "/Bulk/DRAGEN WGS/ML-corrected DRAGEN population level WGS variants, pVCF format [500k release]/chr11" | headUnfortunately the official documentation is no help here. The best I found was a two-year-old community forum reply suggesting each batch covers roughly 20,000 bp. Thanks, George F. You saved me more time than UKB’s own docs did. Take the estimate as a ballpark, not a guarantee.
Here is the approach in practice. To find positions 47,331,406 - 47,352,702 on chromosome 11, you can divide 47,331,406 by 20,000 to derive batch 2366 as a reasonable first guess. I wrote a small script (firstpos.sh) to check the first position of any candidate file:
#!/bin/bash
CHR=$1
BATCH=$2
FILE="/Bulk/DRAGEN WGS/ML-corrected DRAGEN population level WGS variants, pVCF format [500k release]/chr${CHR}/ukb24311_c${CHR}_b${BATCH}_v1.vcf.gz"
URL=$(dx make_download_url "$FILE" --duration 1h)
bcftools query -f '%POS\n' "$URL" | head -1bash firstpos.sh 11 2366returns 47,319,031. This is just below my target region, which is a good sign.
Checking the next batch:
bash firstpos.sh 11 2367returns 47,339,029, meaning batch 2366 covers 47,319,031 - 47,339,028. My region ends at 47,352,702, so I check batch 2368 to find where that’s covered. Once you’ve identified which batches bracket your full region, you have your files. If the 20,000 bp estimate puts you nowhere near the right chromosome positions, jump by a larger increment and binary search from there. Eventually you’ll find your file. Then comes the next question: what do you do with it?
What’s Actually in These Files?
A VCF is a giant matrix. Lines starting with # are metadata: pipeline details, descriptions for variant annotations in the INFO column, chromosome descriptions, and so on. The last # line is the column header for the data rows below it. Each data row is one variant. The first nine columns describe that variant: chromosome, position, ID, reference allele, alternate allele, quality score, filter status, info fields, and format. Everything after column nine is per-sample genotype data. At biobank scale that’s 500,000 columns. The good news is that bcftools can filter by position without loading the entire matrix into memory, which is the whole reason streaming works at scale.
Code to Stream Genetic Data in UK Biobank
That’s exactly what the --regions flag does.
FILE="/Bulk/DRAGEN WGS/ML-corrected DRAGEN population level WGS variants, pVCF format [500k release]/chr11/ukb24311_c11_b2366_v1.vcf.gz"
URL=$(dx make_download_url "$FILE" --duration 1h)
bcftools view "$URL" \
--regions 11:47331406-47352702 \
-O z -o my_region.vcf.gzBcftools reads only what it needs and stops. That’s the whole game: 500,000 participants, one small region, no downloading required.
The Short Version
The release timeline is worth knowing for reading older papers, but you don’t need to memorize it. For most analyses, start with the DRAGEN WGS release (field ID 24311). If your analysis requires haplotype structure, reach for the phased release (field ID 30108) instead, or use both if you need rare variant calls in haplotype context. Once you’ve found the right batch file, dx make_download_url, hands bcftools a URL to a file that may be half a terabyte in size and lets it take only what it needs.
In the next post, I’ll walk through how All of Us handles its genetic data. Spoiler: the documentation isn’t better.
References
Footnotes
Bycroft, C., Freeman, C., Petkova, D. et al. (2018). The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 https://doi.org/10.1038/s41586-018-0579-z ↩
Van Hout, C. V., Tachmazidou, I., Backman, J. D., Hoffman, J. X., Ye, B., Pandey, A. K., et al. (2019). Whole exome sequencing and characterization of coding variation in 49,960 individuals in the UK Biobank. BioRxiv, 572347, https://doi.org/10.1101/572347 ↩
Szustakowski, J. D., Balasubramanian, S., Kvikstad, E., Khalid, S., Bronson, P. G., Sasson, A., … & Reid, J. G. (2021). Advancing human genetics research and drug discovery through exome sequencing of the UK Biobank. Nature genetics, 53(7), 942-948. ↩
Backman, J.D., Li, A.H., Marcketta, A. et al. (2021). Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 599, 628–634, https://doi.org/10.1038/s41586-021-04103-z ↩
Halldorsson, B.V., Eggertsson, H.P., Moore, K.H.S. et al. (2022). The sequences of 150,119 genomes in the UK Biobank. Nature 607, 732–740, https://doi.org/10.1038/s41586-022-04965-x ↩